-
all · 2026Year57Moon16Day
蛋白组学
Read More -
 all · 2026Year59Moon3Day
all · 2026Year59Moon3Day移液工作站和手动移液区别
Read More -
article · 2026Year44Moon1Day
OT-2移液机器人值得买吗?
Read More

Molecular cloning is at the core of synthetic biology as it involves the assembly of DNA and its expression in a target host. However, currently cloning is often a manual, time-consuming and repetitive process, and automation would greatly facilitate this process. Automation of the complete rational cloning process (i.e., from DNA creation to expression in the target host) involves the integration of different operations and machines. There are few examples of such workflows, especially when the design is sound (i.e. the DNA sequence design is fixed rather than based on a random library) and the target host is not genetically tractable (e.g. not sensitive to heat shock transformation) . In this study, an automated workflow is presented for the rational construction of plasmids and their subsequent conjugative transfer into the biotechnological platform organism Corynebacterium glutamicum. The entire workflow is equipped with customized software tools. As an application example, a rationally designed transcription factor biosensor library based on the regulator Lrp was constructed and characterized. A sensor with improved dynamic range was obtained, and insights gained from the screen provide evidence for a dual regulatory function of C. glutamicum Lrp.
Introduction Microbial production of bulk and fine chemicals is an important component in achieving a more sustainable global economy. To facilitate this development, it is necessary to improve the fundamental understanding of microbial life and its engineering to meet society's needs. In recent years, the adoption of the Design-Build-Test-Learn (DBTL) cycle has been proposed as a tool to achieve this goal. (1) Molecular cloning plays an important role in this cycle as it can generate new genotypes with different properties for exploration. A variety of software tools have been developed to assist in the in silico cloning of genotypes, the number of which easily exceeds the number of manual cloning in the laboratory. (2) It is now possible to design hundreds of genotypes in a short time. For example, a production pathway consisting of 5 genes, each with 3 different ribosome binding sites (RBS), has generated 3 5 = 243 variants. However, the design of such a project is relatively simple and the bottleneck now shifts to the actual in vitro creation of these sequences and their expression in the desired industrial host. (3) This problem can be solved by using a one-pot assembly and screening approach, i.e., obtaining many, but not necessarily all, variants and using screening experiments to select the best performing variants. (4) However, these approaches have a drawback: knowledge comes only from a few variants that are successfully constructed and isolated from a one-pot reaction. Variants that are more difficult to construct are less likely to be present in this reaction, making it less likely that their properties will be screened out. For many fundamental biological problems, this is not an ideal solution. Here, rational strain construction and complete traceability of all species within all steps of the workflow are necessary, as is achieved with classical molecular cloning. However, currently molecular cloning is often a manual, time-consuming and repetitive process for which automation would bring huge benefits. (5) Automation of rational cloning workflows has multiple benefits. The hands-on time experimenters spend on strain construction can be significantly reduced. Coupled with the higher throughput that can be achieved, this greatly increases the number of constructs that can be produced in a timely manner. Furthermore, automation introduces standardization into the process by eliminating random variations in the process. These processes can also be more easily monitored and analyzed, making it easier to find room for improvement. Therefore, automation can improve the quantity and quality of cloning workflows. In recent years, microfluidic technology has been used to automate the cloning process. (6,7) Here, a custom microfluidic chip is used to provide liquid separation and liquid transfer for a single unit operation. While this technology has the advantage of combining many unit operations in a single device and being able to scale to a large number of experiments per chip, it requires highly specialized infrastructure and personnel to fabricate the chips and conduct the experiments. A more modular and convenient solution is to use a standard liquid handling system, with auxiliary equipment if required. Such systems can be quite complex, capable of performing a large number of experiments with different tasks, (8−11) and there are more cost-effective solutions available. (12) Recently proposed strategies exploit the natural transformation capabilities of specific bacteria, resulting in an efficient and easy-to-use cloning method. (13) However, this approach is limited to a small number of organisms that actively take up exogenous DNA. Most automated cloning workflows published to date focus on E. coli . (14) Escherichia coli is a mature host for molecular cloning and is very easy to manipulate genetically. Many genetic tools have been developed and optimized for E. coli and comprehensive omics data are available. However, E. coli is not always an ideal host for industrial processes due to its low pressure tolerance and risk of phage infection. (15) Therefore, it would be beneficial to expand automated platforms to encompass engineering other genetically more difficult microorganisms. Corynebacterium glutamicum is a widely used industrial bacterium (16) that is more difficult to engineer than E. coli due to its resistance to automation-friendly transformation processes such as heat shock transformation. Although some steps have been taken in this direction, (17) they often fail to automate the actual target biotransformation process, most commonly because electroporation (the preferred method for such organisms) is not easily automated. This study presents a complete workflow for automated rational construction of heat shock-resistant microbial strains. All work was performed using standard liquid handling systems. PCR and Gibson assembly were used to construct a library of 96 plasmids. An automated protocol for heat shock transformation of E. coli was developed as a shuttle system. Colony PCR and sequencing were used as quality controls. For the final step in transforming C. glutamicum, a novel high-throughput conjugation workflow was developed. Conjugation is a well-described and highly relevant bacterial transformation technique. (18) This method is highly efficient, but is often laborious as it requires the use of agar plates and filter paper. In this study, we simplified and automated the procedure by using centrifugation. The entire workflow is equipped with a custom software tool that tracks all constructs and their status in the process. As an application example, the assembly and expression of different Lrp biosensor variants in Corynebacterium glutamicum are shown. The Lrp biosensor has been previously developed for the detection of l -methionine and branched-chain amino acids in Corynebacterium glutamicum. (19) The sensor combines intracellular l -methionine and branched-chain amino acid concentrations with the expression of eyfp, which encodes a fluorescent reporter protein. Increased intracellular concentration results in enhanced fluorescence signal. In general, the design of biosensors is relatively modular; their properties can be changed by modifying ribosome binding sites, promoter length, etc. (20,21) Furthermore, by design, they provide a direct and easily measurable relationship between genotype and phenotype; i.e., changes in Lrp sensor design may result in different measurable fluorescence outputs. Therefore, rational designs of different Lrp biosensors were chosen as application examples to demonstrate the advantages of our cloning automation approach.
Results and Discussion Automated Genetic Engineering Workflow The automated cloning workflow developed in this study (Fig. 1) can be divided into two stages: assembly and amplification of the plasmid in E. coli and transfer of the plasmid to the target organism (in this case Corynebacterium glutamicum). Plasmids were constructed by in silico design of components, construction of fragments by PCR, and integration into backbone vectors by Gibson assembly. Gibson assembly was chosen because it allows scarless assembly of plasmids. (22) Subsequent transformation into E. coli via heat shock. The resulting clones were cryopreserved and a first step of quality control was performed by colony PCR. This method was chosen due to its cost-effectiveness and short run time. The results of this colony PCR influence the next steps: only E. coli clones with positive colony PCR results, i.e. the resulting DNA fragments are of the expected size, indicating successful assembly of the fragments into the backbone, will be considered for automated plasmid preparation , and simultaneously incorporated into Corynebacterium glutamicum. Purified plasmids were used for sequencing as a final quality control. Afterwards, the automatically constructed strains were screened to characterize altered properties. Best of all, each unit operation is designed to be independent and therefore can be used outside of the workflow.

Automated unit operations are performed or supported by a low-cost liquid handling device, the Opentrons OT-2, and a more sophisticated liquid handling platform based on the previously described Tecan EVO 200. (23,24) The two systems are fundamentally different: OT-2 uses two air-displacement pipettes with disposable tips. The range of available pipette capacities is similar to that of manual pipettes, and the operator must select a pipette suitable for the experiment. It has no manipulator; therefore, it cannot change the position of the plate, for example, by placing the plate on a heating block or in a centrifuge. The platform can accommodate up to nine microtiter plates. The model used in this study did not come with the option to cool labware. A custom cooling rack (see Supporting Information) that can be filled with ice and connector adapters was designed and 3D printed to cool the reagents when necessary (e.g., for Gibson assembly). The EVO 200 in the configuration used in this study has a liquid-based liquid handling system with volumes ranging from 3 to 990 μL. It is equipped with a centrifuge, a cooling carrier and a heater/vibration unit, both available to the integrated robotic manipulator arm. Depending on the carrier used, the platform can accommodate 15 or more microtiter plates. To spot bacterial cultures onto 100 mm round Petri dishes, we designed and 3D printed a custom adapter (see Supporting Information). The decision to use two different liquid handling systems was driven by the need for the most efficient use of laboratory equipment: whenever possible, use the OT-2 for unit operations. This saves time compared to the more expensive EVO 200 and can therefore be used for more demanding experiments. In this study, the unit operation “colony picking” was performed manually. It could have been done automatically with specialized equipment, but these were not available at the time. Rational combinatorial design of transcription factor-based biosensors To demonstrate the applicability of our workflow, different versions of Lrp biosensors were designed, constructed, and expressed in C. glutamicum . The Lrp biosensor combines intracellular l -methionine, l -leucine, l -isoleucine, and l -valine concentrations with the expression of the fluorescent reporter protein eYFP. To construct different versions of the Lrp biosensor, different parts of the sensor were modified. The Lrp biosensor is composed of the lrp gene (encoding the Lrp transcription factor), the divergently expressed eyfp gene (encoding the fluorescent reporter gene), and the intergenic region (including the lrp promoter and the brnF promoter upstream of eyfp and the eyfp RBS) (19) (Fig. 2) Different variants were designed for the lrp start codon and RBS, brnF promoter and eyfp RBS.
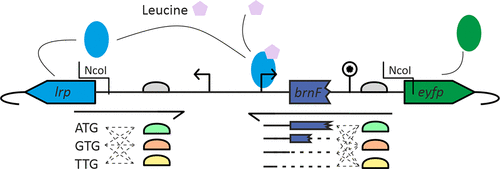
For the lrp start codon, three variants were selected: the natural start codon ATG (should lead to the highest expression), the start codon GTG (should lead to lower expression) and the TGT (should lead to very low or no expression) . Each of these start codon variants was combined with three different lrp RBS, again from highest to lowest expression: GCTAAAATGG (strongest), GCTATTGTGC (native, weaker), and CAATCCTACC (weakest). The combination of three start codons and three RBSs on the lrp side results in 3 × 3 = 9 different primers. The eyfp gene is expressed under the control of the brnF promoter, which contains a binding site for the Lrp protein. (25) Therefore, when the promoter sequence is shortened or lengthened, binding sites can be added or removed. Since brnF is transcribed as a leaderless transcript and the first part of brnF may have a regulatory function, (26) the eyfp promoter of the standard Lrp sensor contains the first 30 bp of brnF. Four different variants were designed in this study, starting from +30 (standard Lrp biosensor promoter) and designing shortened versions in steps of 15, namely +15, 0 and −15. For the RBS of eyfp, three variants were selected: AAAAGGAGAT (strongest), AGAAGGAGAT (native, less strong), and ATCCGACCAT (weakest). The combination of four promoter lengths and three RBSs on the eyfp side resulted in 4 × 3 = 12 different primers. This design strategy includes a total of 9 × 12 = 108 different Lrp biosensor variants. Each variant can be constructed in a single PCR reaction, and all PCR reactions can use the same PCR template, plasmid pJC1- lrp - brnF′ - eyfp. To assemble the biosensor plasmid, each PCR reaction was followed by a two-fragment Gibson assembly, in which the PCR products were assembled into the pEC-Tmob18-lrp-eyfp backbone; all PCR products were designed so that they could be assembled into the same backbone. in the chain.
DNA Assembly and Transformation of E. coli Prior to construction of Lrp biosensor variants, backbone plasmids were constructed by inserting lrp and eyfp into the removable pEC-Tmob18 backbone. A short spacer containing two NcoI restriction sites was inserted between the two genes. This sequence allows the linearization of the plasmid and the insertion in the next step of sequence variants containing different versions of the lrp start codon, lrp RBS, brnF promoter length, and eyfp RBS. Of the 108 variants, 96 were selected as application examples for automated cloning workflows because most automated devices are designed for 96-well multititer plates. PCR variant construction, Gibson assembly, and E. coli heat shock transformation were performed sequentially. The Opentrons OT-2 is capable of automatically pipetting 96 PCR reactions in 85 minutes, followed by manual transfer of the plates to the thermal cycler. After thermal cycling, 96 Gibson assembly reactions were mixed using OT-2, which took 50 min. Incubate by manually transferring the plate to the thermal cycler. Proper cooling of the Gibson master mix before the 50 °C incubation step is critical; otherwise, E. coli transformants will not be obtained. In our workflow, cooling is achieved by placing all labware into small 3D-printed boxes filled with ice. Heat shock transformation of E. coli was performed on a TECAN EVO200 liquid handling robot, which enables a completely hands-free process, starting with Gibson assembly mix and competent cells and ending with spotting the cells on agar plates ready for overnight incubation. The process took 170 minutes. In total, PCR, Gibson assembly, and transformation of 96 constructs were completed within 8 hours, requiring only 40 minutes of hands-on time. A suitable way to implement the heat shock step is to preheat the V-bottom 96-well plate on a heating unit, transfer the 8 cell-plasmid mixtures one at a time from the cooling carrier to the heating plate, incubate for 30 seconds, and then transfer Return to cooling carrier. While it is faster to transfer the entire plate from the cooling carrier to the heating device, this produces less reliable heat transfer results. The cell suspension was concentrated by centrifugation, resuspended in LB medium, and spotted on agar plates by pipetting six 5 μL spots in the same row on a single agar plate. Furthermore, the parameters of heat shock transformation were tested using E. coli competent cells (NEB 5-alpha competent E. coli) and the pUC19 standard vector. Heat shock temperature (37, 42, and 47 °C) and heat shock duration (0 to 30 s in steps of 5 s) were tested. Surprisingly, there was little difference in colony-forming units with successful transformation under each condition. Ultimately, the conditions most similar to standard manual procedures were selected.


The experienced service team and strong production support team provide customers with worry-free order services.

简体中文

繁體中文

English
日本語
한국인