Detection and identification of SARS-CoV-2 lineage B.1.526 in New York
Large-scale sequencing of the SARS-CoV-2 genome is critical to tracking the evolution of the virus during the current outbreak. Variants first discovered in the United Kingdom, South Africa and Brazil have spread to multiple countries. We developed the software tool “Variant Database (VDB)” to quickly examine changes in spike mutations. Using the VDB, we identified an emerging SARS-CoV-2 lineage in the New York region that shares mutations with previously reported variants. The most common set of spike mutations in this lineage (now designated B.1.526) are L5F, T95I, D253G, E484K or S477N, D614G, and A701V. This lineage was first sequenced in late November 2020, when it comprised less than 1% of sequenced coronavirus genomes collected in New York City (NYC). As of February 2021, the genomes of this lineage accounted for approximately 32% of the 3,288 sequenced genomes from New York City specimens. Phylogenetic inference confirmed the rapid growth of the B.1.526 lineage in New York City, particularly the subclade defined by the spike mutation E484K, which outpaced the growth of other variants in New York City. Pseudovirus neutralization experiments showed that the B.1.526 spike mutation adversely affects neutralization titers in convalescent and vaccine recipient plasma, demonstrating the public health importance of this lineage.
Introduction Following the initial months of the 2020 SARS-CoV-2 pandemic, the vast majority of sequenced genomes contained the spike mutation D614G (along with 3 independent nucleotide changes) 1 . After a period of gradual changes, several variants containing multiple mutations emerged in Q4 2020, many of which were located within the spike gene 2 – 5 . Multiple lines of evidence suggest that evasion of antibody selection pressure is a driving force in the development of these variants 6 – 9 .
Genomic surveillance of SARS-CoV-2 is currently focused on monitoring the emergence of these variants and the functional impact their mutations may have on the effectiveness of passive antibody therapies and vaccines in preventing mild or moderate COVID-19. Although more and more samples are being sequenced, analysis of these genomes remains a challenge10. Here, we develop a simple and fast utility to quickly examine the mutational landscape revealed by SARS-CoV-2 genome surveillance: the variant database (vdb). Using this tool, we discovered several recently sequenced genomes that harbor mutations at key antibody epitopes. Within this group is a new lineage that emerged in New York City and has increased in frequency, accounting for approximately 32% of sequenced genomes as of February 2021. We confirmed the rapid spread of B.1.526 in New York City in early 2021 through phylogenetic inference. Additionally, we evaluated the impact of the B.1.526 spike mutation on plasma neutralization titers during the convalescent period and in vaccine recipients.
Results Phylogenetic analysis is critical to understanding the relationships among viral genomes. However, other perspectives are also helpful in detecting patterns in large sequences. We developed vdb as a utility to query sets of spike mutations observed during genome surveillance. Using the vdb tool to analyze SARS-CoV-2 sequences in the Global Initiative for Sharing Avian Influenza Data (GISAID) dataset11,12, we detected several variants that are different from B.1.1.7, B.1.351, and B.1.1.248 and B.1.429 2 – 5 sequence clusters with spike mutations at sites known to be associated with anti-SARS-CoV-2 antibodies8,13 (Table 1). The vdb program can find clusters of viruses with identical sets of spike mutations, and these patterns can then be used to find potentially related sequences.
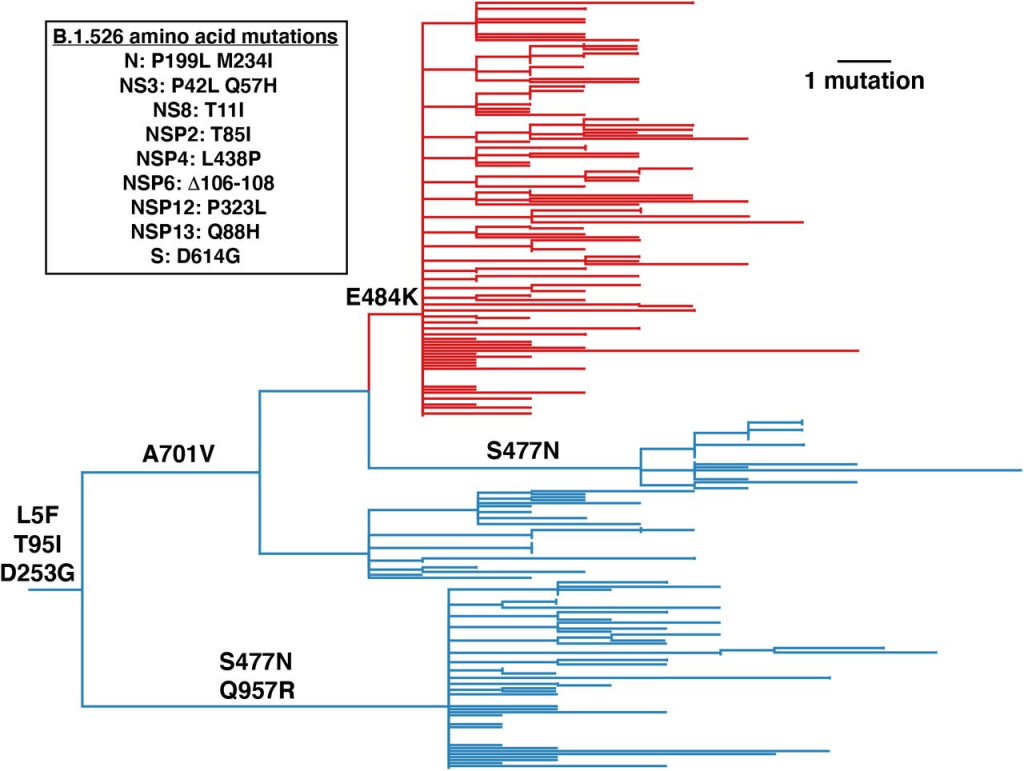
Mutations that define B.1.526 A noteworthy cluster of genomic sequences was collected from the New York region, representing a unique lineage now designated B.1.526 (Fig. 1, Supplementary Fig. 1). This variant is present in clade 20.C and is characterized by 3 defined spike mutations: L5F, T95I, and D253G. In B.1.526, the largest subclade is defined by E484K and two distinct subclades are each defined by S477N; both mutations are located within the receptor-binding domain (RBD) of the spike (Fig. 2 and Supplementary Table 1 ). We note that the evolutionary history of spike position 701 depends on whether the tree is rooted using the molecular clock (Fig. 1) or its sister clade (characterized by the L452R mutation; Supplementary Fig. 2), the latter hypothesized to replace A701V followed by reversion to V701A . Among the nucleotide mutations in the B.1.526 lineage, the most characterized include A16500C (NSP13 Q88H), A22320G (spike D253G), and T9867C (NSP4_L438P). Another striking feature of the B.1.526 lineage is the deletion of nucleotides 11288-11296 (NSP6 106-108), which is also present in variants B.1.1.7, B.1.351, P.1 and B.1.525 14 in.
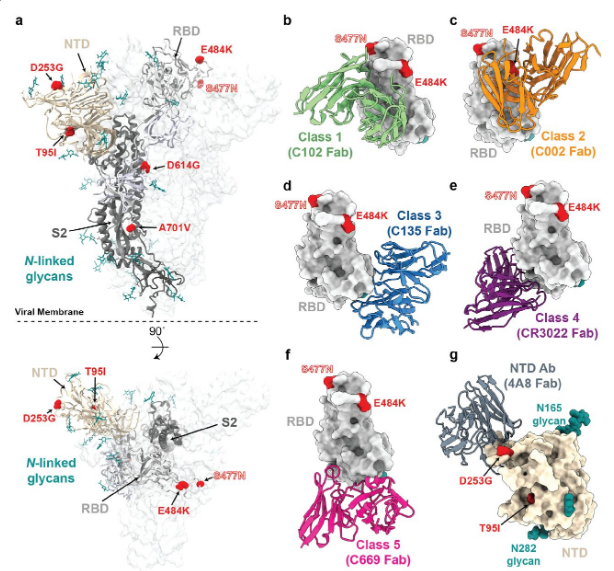
Figure 1 Phylogenetic tree of lineage B.1.526 showing spike mutations. Maximum likelihood phylogeny of SARS-CoV-2 variant B.1.526 sampled by NYC PHL (n=258). Spike protein amino acid substitutions occurring on the inner branch are labeled, including three spike mutations unique to B.1.526. The B.1.526 clade defined by the E484K mutation is highlighted in red. Inset highlights non-spike amino acid substitutions and deletions that distinguish the B.1.526 clade from the Hu-1 reference genome. For display purposes, only the NYC PHL genome is shown.Figure 2.B.1.526 Structural location of lineage spike mutations. a, Side and top views of the SARS-CoV-2 spike trimer (PDB 7JJI), with mutations of lineage B.1.526 shown as spheres. bg , representative neutralizing antibody models (cartoon, VH-VL domains only) bound to the RBD (bf, gray surface) or NTD (g, wheat surface). Sites mutated in the B.1.526 lineage are shown as red spheres. Also shown is the S477N site of the branch containing this mutation instead of the E484K mutation (see Figure 1); b, category 1 (PDB 7K8M); c, category 2 (PDB 7K8S); d, category 3 (PDB 7K8Z ); e, Class 4 (PDB 6W41); f, Class 5 8; g, NTD-specific antibodies 4A8 (PDB 7C2L).
Regarding the four spike mutations prevalent in this lineage: (1) E484K is known to attenuate the neutralizing effect of multiple anti-SARS-CoV-2 antibodies, particularly those found in class 2 anti-RBD neutralizing antibodies13, 15 , and is also present in variants B.1.351 4 and P.1/B.1.1.248 2 , (2) D253G is reported to be targeted against N An escape mutation of terminal domain antibodies16, (3) S477N has been identified in several early lineages17, is close to the epitope of multiple antibodies18, and has been shown to increase viral infectivity through enhanced interaction with ACE2 19 , 20 , and (4) A701V is located near the S2′ cleavage site of the adjacent protomer and is shared with variant B.1.351 4 . The overall mutation pattern of the B.1.526 lineage (Fig. 2 ) suggests that this lineage arose in part due to selective pressure from antibodies. Based on the dates these viruses were collected, it appears that this lineage is rapidly increasing in frequency in New York.
 article · 2025Year47Moon25Day
article · 2025Year47Moon25Day article · 2025Year34Moon25Day
article · 2025Year34Moon25Day article · 2025Year21Moon24Day
article · 2025Year21Moon24Day

